

Abstract
Introduction:
Diffuse large B cell lymphoma (DLBCL) is a clinically heterogeneous disease. While roughly half of the patients respond well to standard R-CHOP therapy, the majority of the remainder succumb to their disease. While many targeted therapies have been developed in DLBCL, resistance to single agents develops almost invariably. While drug combinations have proved to be effective approaches to overcoming resistance in infectious diseases, developing such combinations has proved to be difficult in diffuse large B cell lymphomas and other cancers owing to not only the heterogeneity of these diseases and overlapping toxicity profiles.
We hypothesized that with advent of powerful new machine learning approaches combined with genomics, that we would be able to identify novel mechanisms of resistance and develop effective combination therapies to overcome resistance to single agents.
Results
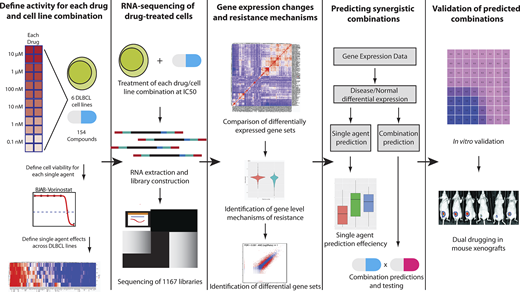
We tested in vitro responses to all FDA-approved and Phase III cancer drugs (N=150) in six DLBCL cell lines carefully chosen to represent the heterogeneity with regard to cell of origin and common genetic alterations. We observed that roughly half of our drugs were active in at least 50% of the DLBCL cell lines.
We then performed RNA sequencing on these cell lines before and after exposure to each of these drugs at their specific IC50 (concentration of drug required to kill 50% of the cells). In addition, we tested the effects of 38 cytokines and antibodies to assess their downstream biological effects. In all, we generated 1167 RNAseq profiles post-exposure to drug (N=900) or cytokines. Hierarchical clustering of our RNAseq data demonstrated clusters of drugs with shared mechanisms and targets (e.g. HDAC inhibitors, PI3K and mTOR inhibitors).
We developed a machine learning approach using a combination of neural networks and Bayesian network propagation analysis to identify pathway activation and mechanisms of resistance associated with each of the drugs. Our approach identified 16 combinations of drugs that had different mechanisms and downstream targets.
Surprisingly, we found that histone deacetylase inhibitors (HDACi, e.g. panobinostat) were predicted to be strongly synergistic in combination with JAK inhibitors (e.g. ruxolitinib). These findings were unexpected as ruxolitinib had very weak single agent effects and the JAK-STAT pathway is not thought to be specifically associated with response to HDACi.
We verified the predictions of the machine learning algorithm by performing in vitro combination assays in six different cell lines. In each case, we found that the combination was highly synergistic using the Chou-Talalay method. We further verified the feasibility and efficacy of combining panobinostat (HDACi) and the JAK inhibitor ruxolitinib in vivo using xenograft models. Both single agents had relatively modest effects on tumor burden, but we found significant synergy with the combination (p<0.01), with vastly decreased tumor burdens. In vivo modeling also allowed for testing for hematological toxicity. Hemoglobin levels and ANC remained constant with all therapies, though combination therapy caused a 25% decrease in platelet levels, which would be considered clinically tolerable with monitoring. We further performed mechanistic experiments that demonstrate that JAK-STAT pathway activation through genetic mutations in STAT3 directly contribute to HDACi resistance and reverse sensitivity to HDACi.
Conclusions
These results provide a powerful proof of principle for the application of large scale perturbation approaches combined with machine learning to identify novel drug combinations and mechanisms of resistance.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal